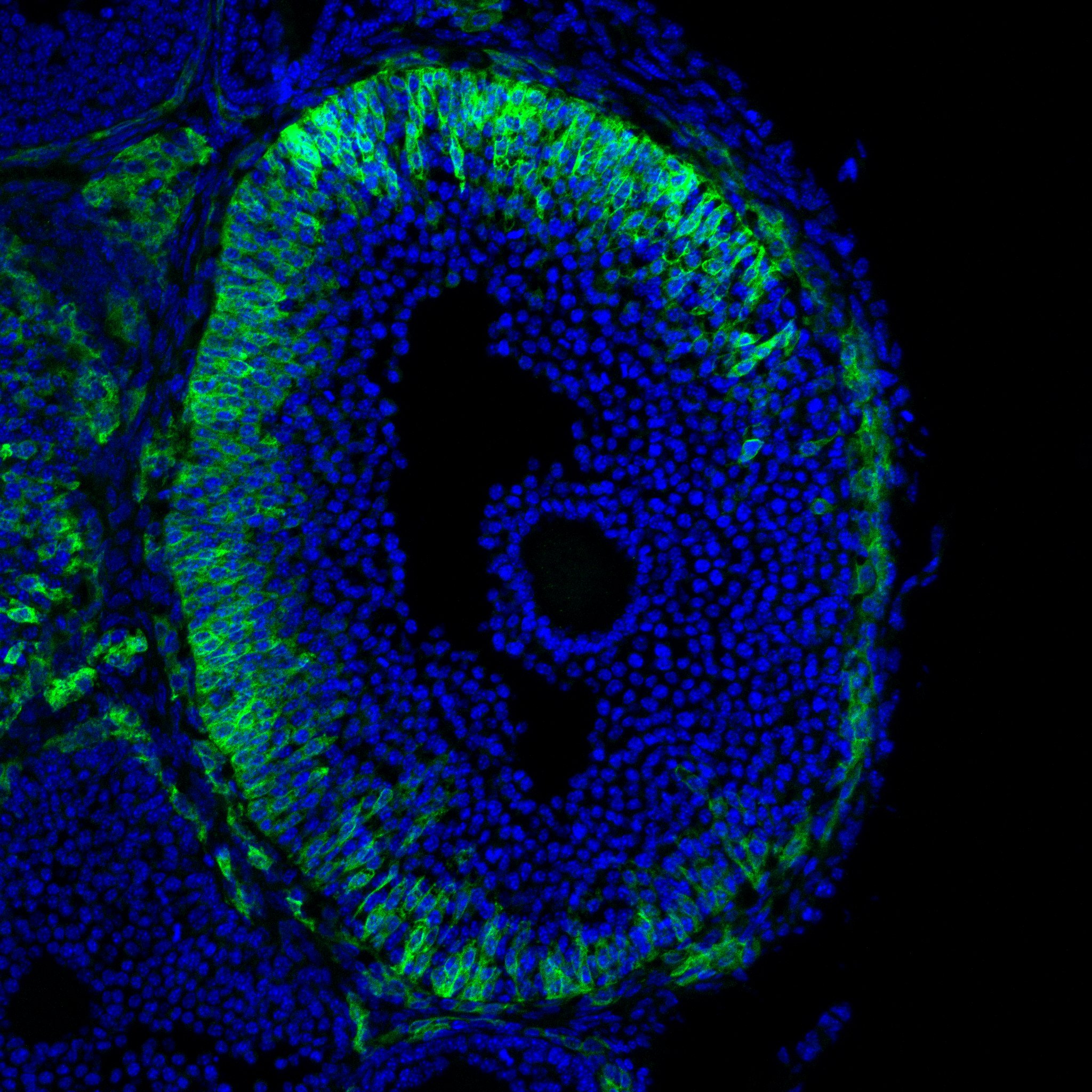
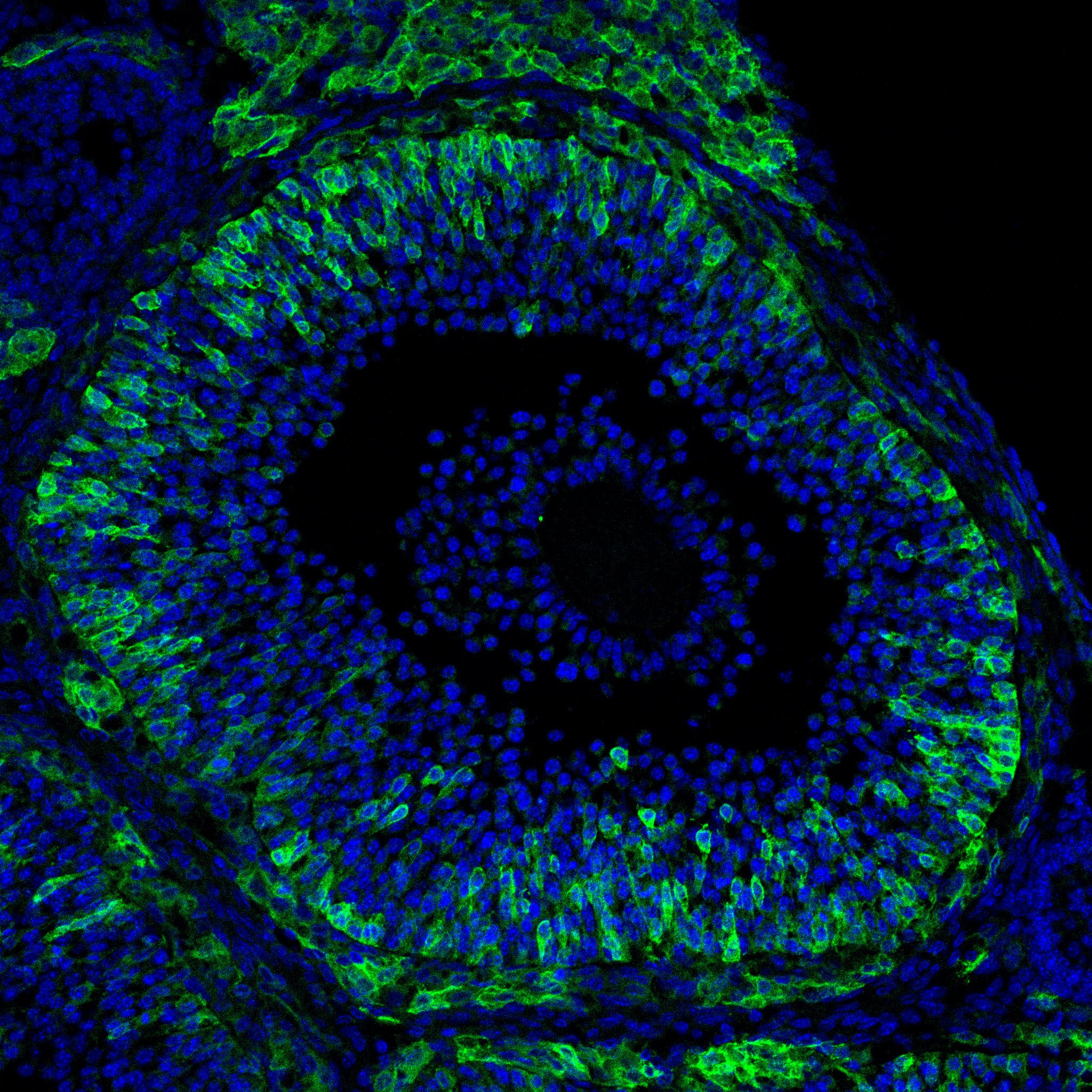
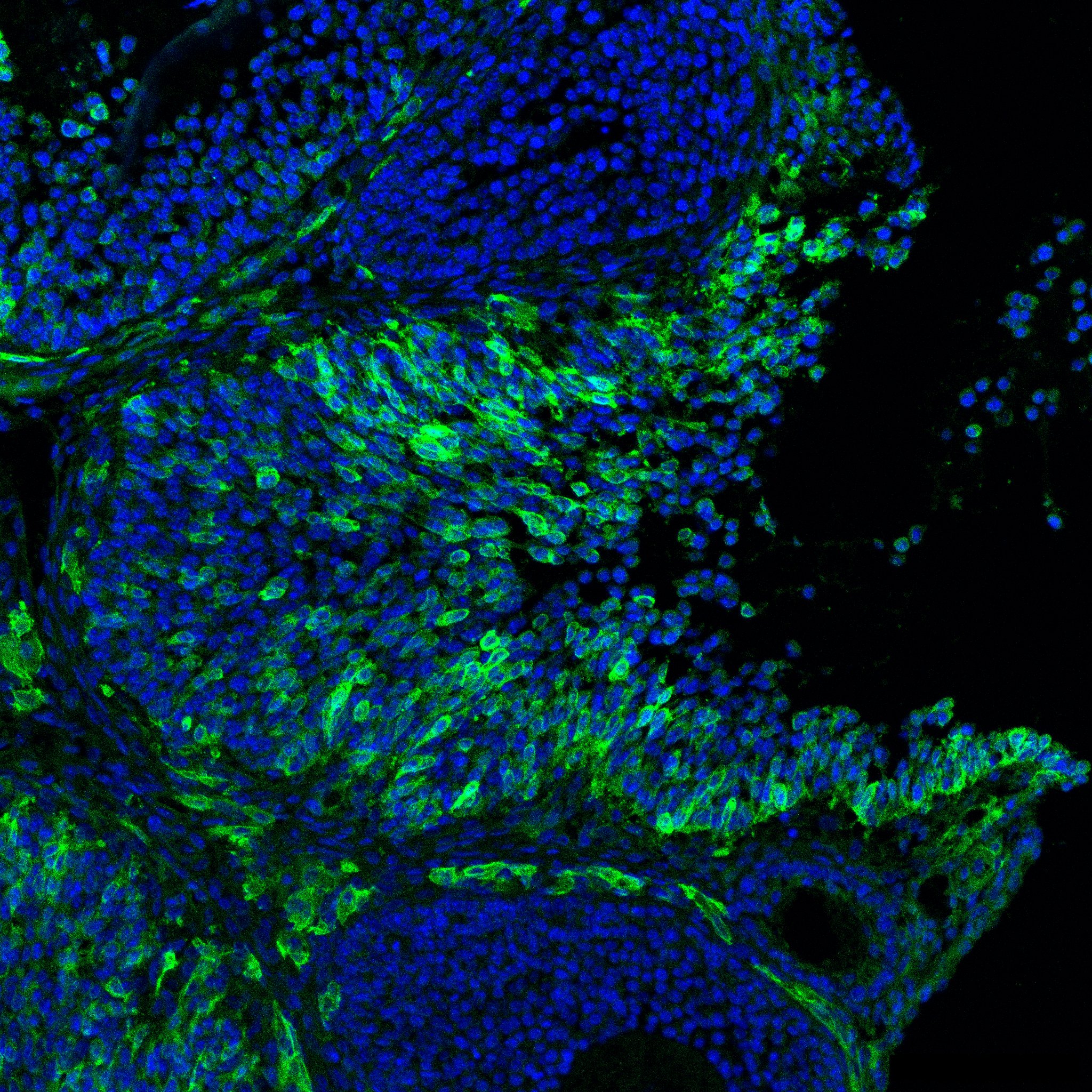
Research in the Department of Cell Biology is concerned with cells as well as how cells function in the context of the various tissues of the body. Our goal is to discover molecular and physiological mechanisms that underlie the treatment and prevention of human disease.
The department faculty participate in the first year medical and dental student curriculum, and the Foundations of Biomedical Science course for graduate students. Several faculty are also contributors to medical student textbooks.
The core of the department consists of former members of the Department of Physiology, which was chaired by Richard D. Berlin for over 30 years. He oversaw the change to Cell Biology, the addition of members from the former Departments of Pharmacology and Anatomy, and the formation of the Center for Vascular Biology (Linda Shapiro, Director) and the Center for Cell Analysis and Modeling (Pedro Mendes, Director). The Department of Cell Biology is also the academic home for basic science faculty members in the Pat and Jim Calhoun Cardiology Center (Christopher Pickett, Interim Director) and the Center for Quantitative Medicine (Pedro Mendes, Director). The department organizes the annual Richard D. Berlin lecture.
Cell Biology Retreat

Cell Biology and Vascular Biology, 2017 retreat at the Pond House (photo by Charan Devarakonda)
Department News
Congratulations to Mayu Inaba and her lab on their publication in Nature Communications!
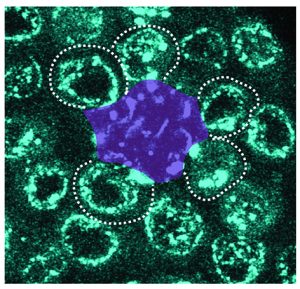
Ridwan, Twillie, Poursaeid, Beard, Bener, Antel, Cowan , Matsuda , and Inaba
Diffusible fraction of niche BMP ligand safeguards stem-cell differentiation
Nature Communications (2024) 15, 116.
UConn Today
Seminars 2024
February 2, 2024
Ian Hitchcock, University of York. "Even better than the real thing? Unlocking the functional pleiotropy of thrombopoietin" (H. Oguro and V. Scanlon, Hosts) Joint seminar with Center of Regenerative Medicine & Skeletal Development. Hybrid
March 6, 2024
Swathi Yadlapalli, University of Michigan. "Structure and organization of circadian clocks" (Inaba Oguro, Host). Hybrid
March 13, 2024
Jamil R. Azzi, Harvard University. “From the bench to the clinic and back” (Terasaki, Host). Hybrid
May 28, 2024
Molly Moravek, University of Michigan. "Mouse models to study reproductive effects of gender-affirming hormones" (Mehlmann, Host). Joint seminar with the Department of Obstetrics and Gynecology. Hybrid
October 1, 2024
Elizabeth Chen, University of Texas Southwestern. Cell Biology Department Retreat.
October 17, 2024
Caleigh Mandel-Brehm, Yale, (Jaffe, Host). Joint seminar with Neuroscience and the graduate student organization. Hybrid
November 13, 2024
Nicole Valenzuela, UCLA (Murphy, Host). Hybrid
December 13, 2024
Tom Seegar, University of Cincinnati (Murphy, Host). Hybrid